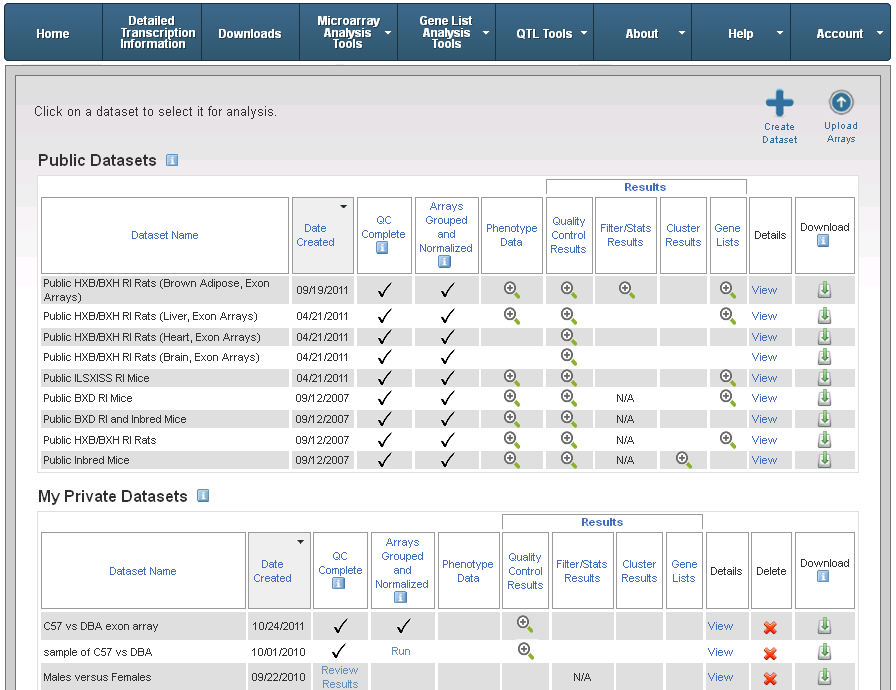
You can see the collection of all your datasets at any time. The page that displays your datasets shows you the stage that each dataset is in (e.g., quality control completed, grouped and normalized, etc.)
At the top of the page, you can click the Create Dataset option if you want to retrieve and select arrays and finalize them into a new dataset, or click the Upload Arrays option to upload your own arrays and create a dataset. The page provides four grouped and normalized "Public" datasets for you to analyze and save new gene lists. See "Public Datasets" for information about public datasets.
Another table displays after the Public Datasets table and shows your "Private" datasets. After you finalize a dataset, it becomes part of the My Private Datasets table, where your progress on that dataset is denoted in the columns:
You can:
The Public datasets available for analysis on the PhenoGen website are pre-compiled groupings of gene expression data for various strains of inbred and recombinant inbred mice and rats. These datasets are available for all types of analysis by any registered users but may be most useful for performing correlation analysis with phenotype data. These datasets are normalized using the most common normalization techniques and have already had quality control checks run. Additionally, datasets created using the Affymetrix Exon arrays have been adjusted for batch effects using an empirical Bayes method (Johnson et al 2007). The normalized data or raw data can be downloaded from the Download Resources page.
The whole brain gene expression dataset for the inbred mice includes 20 inbred strains. Each strain has four to seven biological replicates for a total of 90 individual arrays. The whole brain mRNA for each naive 10-12 week old male mouse was hybridized to a separate array, i.e., no pooling of samples.
The inbred mouse data was normalized nine different ways. Five of the normalization methods are available on the website. For the other four versions, a probe mask was created to eliminate probes whose sequences did not match to the NCBI m37 Build, matched the genome in multiple places, or harbored a SNP between any of the 19 strains where genotype data is available at the Imputed Genotype Resource from the Jackson Laboratory; http://cgd.jax.org/datasets/popgen/imputed.shtml (129P3/J is not available). Entire probe sets were eliminated if less than four associated probes remained. The version using the probe mask and the RMA normalization method is the RECOMMENDED version.
The whole brain gene expression dataset for the BXD recombinant inbred mice includes 30 recombinant inbred strains and the two parental strains (C57BL/6J and DBA/2J). Each strain has four to seven biological replicates for a total of 172 individual arrays. The whole brain mRNA for each naive 10-12 week old male mouse was hybridized to a separate array, i.e., no pooling of samples.
The BXD data was normalized nine different ways. Five of the normalization methods use all of the probes in the dataset. A probe mask was created for the other four versions to eliminate probes whose sequences did not match to the NCBI m37 Build, matched the genome in multiple places, or harbored a SNP between any of the 19 inbred mouse strains included in the public dataset (according to Imputed Genotype Resource from the Jackson Laboratory; http://cgd.jax.org/datasets/popgen/imputed.shtml). Entire probe sets were eliminated if less than four associated probes were eliminated. The version using the probe mask and the RMA normalization method is the RECOMMENDED version.
For the eQTL analysis of this data set, a slightly different mask was used. Instead of eliminated probes with SNPs between the 19 inbred strains, probes were eliminated if they contained a known SNP between the two BXD parental strains, based on whole genome sequence data from the Sanger Institute (Keane et al 2011). Expression values were normalized and summarized into probe sets using RMA. MAS5 was used to evaluate if expression level measurements were above background noise (present, absent, or marginal). If a probe set did not have at least one present call throughout all samples, the probe set was dropped from the data set. Of the 41,581 probe sets retained after masking, 30,031 probe sets remained after filtering by present/absent calls. Data were thoroughly examined for batch effects related to processing. The microarrays were run over a year and a half period, resulting in 15 batches. Both batches and strains contribute to non-random data distribution and a new method for removing batch effects, while retaining strain effects, was used (personal communication, Evan Johnson, Boston University) on the set of 30,031 probe sets detected above background. This method combines a simple rank test and a Bayesian hierarchical framework similar to the empirical Bayes method, Combating Batch Effects When Combining Batches of Gene Expression Microarray Data (ComBat) (Johnson et al., 2007). This version of the data is available in the Download Resources section.
This expression data set represents a combination of the two datasets previously mentioned. In this dataset there are a total of 50 strains (C57BL/6J and DBA/2J are in both of the previous sets) and 253 individual arrays. See the preceding topic for details on "masked" versions.
The whole brain gene expression dataset for the LXS recombinant inbred male mice includes 59 recombinant inbred strains (one strain (LXS49) was eliminated due to unresolved questions about true strain origin) and two parental strains (ILS and ISS). Each strain has three to six biological replicates, for a total of 342 individual arrays that passed quality control standards. In addition, to control for batch effects, C57BL/6J mice were hybridized to arrays and included in every batch (35 individual arrays), and DBA/2J mice were included in a few of the final batches (9 arrays). The whole brain mRNA for each naive 10-12 week old male mouse was hybridized to a separate Affymetrix Mouse Exon Array 1.0 ST, i.e., no pooling of samples.
Individual probes were eliminated prior to normalization if their sequence did not match any part of the NCBI m37 Build of the mouse genome, if their sequence matched multiple locations in the mouse genome, or if the location in the genome that the probe did match contain a SNP between any of the 19 strains in the public Inbred Mice dataset where genotype data is available at the Imputed Genotype Resource from the Jackson Laboratory; http://cgd.jax.org/datasets/popgen/imputed.shtml (same mask that is implemented on PhenoGen). Entire probe sets were eliminated if less than three of the original probes remained after filtering. Arrays were examined for quality, and arrays that did not meet quality standards were eliminated.
Data from individual probes was normalized using RMA and summarized either into the full set of transcript cluster or the core set of transcript clusters. In addition, RMA values for the full set of individual probe sets is available for download from the resource page, but is not available for analysis on PhenoGen at this time.
Each data set was adjusted for batch effects using the empirical Bayes method outlined by Johnson et al (2007). After batch effects adjustment, C57BL/6J and DBA/2J arrays were dropped from the data set. The version using the probe mask and the RMA normalization method on the core transcript clusters is the recommended version and was used for calculation of eQTLs.
The whole brain gene expression dataset for the HxB/BxH recombinant inbred rats on the CodeLink Whole Genome Rat Array includes data from 26 recombinant inbred strains, the two parental strains (SHR/Ola and BN-Lx/Cub), and the SHR-Lx/Cub strain. The whole brain mRNA of four to seven naive 12-14 week old male rats from each strain were hybridized to separate CodeLink Whole Genome rat arrays (one rat per array) for a total of 139 arrays.
In addition to the five normalization versions available on the website, an "eQTL version" of the dataset that was used for all HXB/BXH rat eQTL calculations is available. This version was obtained by first removing probes from the datasets if they were one of the negative or positive controls placed on the array by the manufacturer. Next, individual values were eliminated based on the quality flags assigned by the CodeLink Expression Analysis Software. Values were eliminated if they were flagged as M (spot was identified to be defective through image inspection at manufacturing), C (spot has a high level of background contamination), I (spot has an irregular shape), or S (spot has a high number of saturated pixels). Values were retained if they were flagged G (spot is good) or L (spot is below local background noise). Also, to be able to take the log base 2 transformation of the background-adjusted intensity values, all background-adjusted intensity values below zero were replaced with the value 0.00001. The data was then normalized using a cyclic LOESS procedure executed in R to account for the missing intensity values.
The HXB/BXH recombinant inbred panel also has four data sets available on transcription levels from the Affymetrix Rat Exon array. Data was collected on whole brain, left ventricle (heart), liver, and brown adipose tissue (BAT) of 21 HXB/BXH RI strains (only 19 RI strains included in the BAT tissue data set) and 6 related inbred strains. Each strain has three to four biological replicates for a total of 108 individual arrays from brain, 105 arrays from heart, 106 arrays from liver, and 96 arrays from brown adipose tissue that passed quality control standards. The mRNA for each naive 10 week old male rat was hybridized to a separate Affymetrix Rat Exon Array 1.0 ST, i.e., no pooling of samples.
Individual probes were eliminated prior to normalization if their sequence did not match any part of the RGSC version 3.2 of the rat genome, if their sequence matched multiple locations in the mouse genome, or if the location in the genome that the probe did match contain a SNP between the Brown Norway (BN/SsNHsdMcwi) inbred strains (reference strain) and the spontaneously hypertensive rat (SHR/OlaIpcv) strain that was recently sequenced (Atanur et al 2010) using next generation sequencing or a SNP detected in DNA sequencing of the BN-Lx/CubPrin and SHR/OlaIpcvPrin strains (same mask that is implemented on PhenoGen). DNA sequence data for the BN/SsNHsdMcwi and SHR/OlaIpcv was downloaded directly from the Ensembl ftp site at: ftp://ftp.ebi.ac.uk/pub/databases/ensembl/snp/rat/shr/.
For the 4,022,111 original probes, 604,601 were removed (472,072 did not map uniquely to the genome; 132,529 probes contained a SNP). Entire probe sets were eliminated if less than three of the original probes remained after filtering. Arrays were examined for quality and arrays that did not meet quality standards were eliminated.
Data from individual probes was normalized using RMA and summarized either into the full set of transcript cluster or the core set of transcript clusters. In addition, RMA values for the full set of individual probe sets is available for download from the resource page, but is not available for analysis on PhenoGen at this time.
Each data set was adjusted for batch effects using the empirical Bayes method outlined by Johnson et al. (2007). The version using the probe mask and the RMA normalization method on the core transcript clusters is the recommended version and was used for calculation of eQTLs.
References
 Take a look
Take a look